轉錄水平解析昆蟲病原線蟲共生菌菌株新型殺菌蛋白PPIA-L20基因表達調(diào)控——材料與方法
1 材料與方法
1.1 試驗材料
伯氏致病桿菌Xenorhabdus bovienii NN6菌株是本實驗室從斯氏線蟲Steinernema oregonense侵染致死的大臘螟血腔中分離鑒定的,菌種保存于實驗室-80 ℃超低溫冰箱中。尖孢鐮刀菌西瓜專化型(Fusariurn oxysporum f. sp. niveum)的1號生理小種,由江蘇省農(nóng)業(yè)科學院蔬菜所瓜類組提供。NBTA鑒別培養(yǎng)基:酵母提取物3 g·L-1,胰蛋白胨5 g·L-1,營養(yǎng)瓊脂(NA)20 g·L-1,2,3,5-三苯基氯化四氮唑(TTC)0.1 g·L-1,溴百里酚藍(BTB)0.025 g·L-1,丙酮酸鈉(SP)1 g·L-1。LB+SP液體培養(yǎng)基:酵母提取物5 g·L-1,胰蛋白胨10 g·L-1,NaCl 10 g·L-1,丙酮酸鈉(SP)1 g·L-1。YSG液體培養(yǎng)基:甘油6.9 g·L-1,大豆蛋白胨25.17 g·L-1,MgSO4·7H2O 1.57 g·L-1,(NH4)2SO4 2.55 g·L-1,KH2PO4 0.87 g·L-1,K2HPO4 1.11 g·L-1,Na2SO4 1.81 g·L-1。馬鈴薯培養(yǎng)基(PDA):馬鈴薯200 g·L-1,葡萄糖20 g·L-1,瓊脂15~18 g·L-1。PSA培養(yǎng)基:馬鈴薯蔗糖培養(yǎng)基粉末46 g·L-1。
1.2 試驗方法
1.2.1 共生菌的活化與發(fā)酵生長曲線的測定
從-80 ℃超低溫冰箱取出菌種在NBTA平板上劃線培養(yǎng),72 h后接種藍綠色單菌落于LB+SP液體培養(yǎng)基中,28 ℃、180 r·min-1活化12 h。將活化好的菌液以5%(體積分數(shù))的接種量加入200 mL YSG培養(yǎng)基(1 L容積錐形瓶),28 ℃、150 r·min-1培養(yǎng)84 h。每4 h取1次樣,每次吸取1 mL菌液加入比色皿中,測定不同培養(yǎng)時間的菌液D600值,菌株生長進入穩(wěn)定期后,每12 h測1次D600值,連續(xù)測量84 h,以時間為橫坐標,菌液的D600值為縱坐標。取各時間段的新鮮菌液1 mL,分別稀釋10、102、103、104、105、106、107,從稀釋好的菌液中吸取200 μL菌液到NBTA培養(yǎng)基,用玻璃棒涂布均勻后倒置于28 ℃培養(yǎng)箱中,待平板長出菌落,開始計數(shù)。每個稀釋濃度重復3次。
1.2.2 胞外粗蛋白的制備
使用硫酸銨沉淀法沉淀粗蛋白,將硫酸銨固體研磨成粉末,加入待沉淀溶液中,至硫酸銨飽和度分別達到10%、20%、30%、40%、50%、60%、70%、80%、90%和100%。4 ℃靜置6 h,12 000 r·min-1離心20 min,得到的蛋白沉淀利用緩沖液復溶后透析除鹽,供后續(xù)試驗使用。
1.2.3 細菌總RNA提取、質(zhì)量檢測和測序文庫構建
分別取1 mL不同發(fā)酵時間的新鮮菌液,加入1.5 mL滅菌離心管中,4 ℃、8 000 r·min-1離心20 min去除上清液。將離心管插入液氮中速凍。每個樣品重復收集3份保存于-80 ℃冰箱。樣品在液氮中充分研磨破壞細菌細胞壁,加入700 μL Trizol充分混合,劇烈振蕩,靜置5~15 min。加入200 μL氯仿(除去蛋白)劇烈振蕩,靜置5 min。4 ℃、12 000 r·min-1離心15 min。小心吸取600 μL上清液(不能吸到中間層蛋白),加入等體積異丙醇(沉淀RNA)緩慢搖勻,-20 ℃靜置30 min。離心機4 ℃預冷,12 000 r·min-1離心5 min,棄上清液。加入400 μL 75%(體積分數(shù))乙醇顛倒4次清洗,12 000 r·min-1離心5 min,棄上清液,離心管傾斜放在超凈臺吹干,每管加入40 μL RNAase free H2O振蕩搖勻,10 000 r·min-1 離心3 min。通過微量分光光度計NanoDrop測定RNA溶液的濃度和純度。將提取的RNA迅速放置液氮中冷凍,委托北京諾禾致源科技股份有限公司完成測序和文庫構建,并使用Illumina測序儀進行測序。
1.2.4 內(nèi)參引物和qPCR引物設計
內(nèi)參基因選擇16S rRNA和ropB,利用Primer-BLAST軟件,設計引物序列。引物由生工生物工程(上海)有限公司合成。內(nèi)參引物序列見表1。
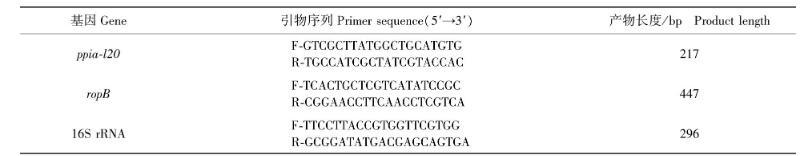
表1 RT-qPCR內(nèi)參引物序列
1.2.5 測序數(shù)據(jù)分析和差異表達基因的篩選
測序數(shù)據(jù)已上傳至National Center Biotechnology Information(NCBI)的SRA數(shù)據(jù)庫(查詢號:PRJNA1047324)。測序平臺為Illumina Novaseq6000,利用Hisat 2軟件將測序數(shù)據(jù)與參考基因組進行序列比對; 采用fastp軟件對原始數(shù)據(jù)進行質(zhì)量預處理,并對整個質(zhì)控過程中的reads進行統(tǒng)計匯總。采用DESeq篩選差異表達基因。
1.2.6 差異表達基因的GO(gene ontology)和KEGG(Kyoto encyclopedia of genes and genomes)富集分析
富集分析采用BLAST2GO和topGOR包進行GO功能分析,得到所有顯著性差異基因、上調(diào)差異基因以及下調(diào)差異基因富集的功能模塊并繪圖; 利用KEGG數(shù)據(jù)庫對差異蛋白編碼基因進行pathway分析(結合KEGG注釋結果),并用超幾何分布檢驗方法計算每個pathway條目中差異基因富集的顯著性。
1.2.7 實時定量PCR分析
選取部分基因通過Real-time PCR對轉錄組結果進行驗證,利用反轉錄試劑盒(ATG巨匠)對1.2.3節(jié)中獲得的RNA進行反轉錄,得到的cDNA采用核酸定量儀Thermo Scientific NANO-Drop1000(NanoDrop Technologies,USA)進行定量,Real-time PCR靶標基因和所用引物見表2。在RNase-free離心管中配制如下混合液:RNase Free ddH2O、4 × gDNA wiper Mix、RNA template混合,用移液器輕輕吹打混勻,42 ℃加熱2 min去除模板中的DNA。5 × ATGScriptTM RT Mix與反應液混合成20 μL反應體系,50 ℃反應15 min,85 ℃反應2 min。每個樣本3個重復。

表2 RT-qPCR引物序列
1.3 數(shù)據(jù)統(tǒng)計與相關性分析
使用Prism 8.0(Graphpad)軟件進行數(shù)據(jù)統(tǒng)計分析,所有統(tǒng)計數(shù)據(jù)均表示為平均值±標準差。采用Student's t-test檢驗, P<0.05被認為具有統(tǒng)計學意義。采用SPSS Statistics 26進行變量間相關性分析。兩變量間相關性以Pearson相關系數(shù)r表示。
相關新聞推薦
1、藜麥和藍靛果酵母菌株篩選、培養(yǎng)、計數(shù)及混菌液態(tài)發(fā)酵工藝優(yōu)化(三)
3、牛肉膏蛋白胨、南極磷蝦蛋白胨微生物培養(yǎng)及生長變化